La Epilepsia es una enfermedad cerebral que se caracteriza por la presencia de crisis epilépticas no provocadas, que se repiten en el tiempo. Afecta a más de 50 millones de personas en el mundo, por lo que ha sido declarada por la OPS-OMS como una patología frecuente, tratable en su mayoría, pero que afecta severamente al individuo en términos de calidad de vida siendo altamente estigmatizadora.
Puede presentarse en cualquier momento de la vida, en cualquier sexo y nivel socioeconómico.
Las causas de la Epilepsia son variables, siendo las más frecuentes a nivel mundial: TEC o traumatismo encéfalo craneano, infecciones del sistema nervioso central (meningitis, encefalitis), problemas en el momento del parto o en el recién nacido (causas perinatales), accidentes cerebro vasculares y causas genéticas.
El diagnóstico de la epilepsia es esencialmente clínico, lo realiza el neurólogo en base a una adecuada historia y examen físico, los exámenes sólo ayudan a precisar y clasificar el tipo de epilepsia (son complementarios).
Las crisis epilépticas son la presentación clínica de la enfermedad, se producen por una descarga eléctrica excesiva a nivel cerebral. El tipo de crisis depende de: lugar del cerebro afectado, velocidad de la descarga epiléptica a través del cerebro, etiología de la epilepsia y la edad de la persona, por lo que no todas las personas con epilepsia, tienen el mismo tipo de crisis epiléptica.
El pronóstico de la epilepsia depende de la causa, así como del inicio temprano del tratamiento y su adherencia. Se estima que hasta el 70% de las personas con epilepsia pueden llevar una vida normal con el tratamiento apropiado, el 30% restante tendrá una epilepsia refractaria, lo que quiere decir, que tendrá crisis a pesar del tratamiento correctamente indicado, en estos casos se plantean otros tipos de tratamiento complementarios como: cirugía de la epilepsia, dieta cetogénica, entre otros.
El uso de fármacos anticrisis será indicado de forma personalizada, de acuerdo con el tipo de crisis, síndrome epiléptico, comorbilidades asociadas, estilo de vida, preferencias personales y/o la relación con su familia y sus cuidadores. Por esto la Epilepsia requiere de un manejo integral.

Una crisis epiléptica se define como la aparición transitoria de signos y/o síntomas debido a una actividad neuronal excesiva o sincrónica en el cerebro.
Estos síntomas, es decir lo que observamos en el paciente o lo que éste relata del evento, dependerán de la región cerebral en donde se produzca la descarga eléctrica anormal.
Las crisis epilépticas provocadas son aquellas que se originan o son consecuencia de una alteración orgánica conocida y aguda. Por ejemplo: la descompensación que sufre una persona alcohólica o adicta en periodo de abstinencia; la fiebre en algunos lactantes, que puede llevarlos a tener una convulsión “febril”; la privación extensa de sueño y la hipoglicemia o disminución importante del nivel de azúcar en la sangre; entre otros.
Las crisis epilépticas no provocadas o espontáneas, que orientan hacia un diagnóstico de Epilepsia, se caracterizan por ser:
· Las crisis epilépticas corresponden al 1% de las consultas en los Servicios de Urgencia de adultos y al 2% de las consultas en pediatría.
· La mayoría de estas crisis, recibidas habitualmente en Servicios de Urgencia, son crisis epilépticas provocadas.
· 1 de cada 10 personas presentará una crisis epiléptica en algún momento de su vida.
· 1 de cada 150 personas en el mundo tendrá Epilepsia, y, por lo tanto, presentará crisis epilépticas no provocadas: espontáneas, estereotipadas y recurrentes en el tiempo.
· Las crisis convulsivas solo afectan al 23% de quienes son diagnosticados con Epilepsia.

En este tipo de crisis se compromete solo una porción del cerebro en su inicio, la crisis es unilateral y comienza en uno de los hemisferios cerebrales. La manifestación clínica de la crisis es diversa y puede presentarse o no compromiso de conciencia en el paciente.
Describiremos aquellas más frecuentes.
La forma más corriente es la sacudida de una parte de una mitad del cuerpo. Las áreas corporales más frecuentes comprometidas son en orden descendente: el rostro, la lengua, las manos y el pie. Ej. Convulsiones en un lado del rostro, giro de la cabeza y de los ojos hacia un lado, posturas mantenidas de parte o todo un hemicuerpo, imposibilidad de hablar.
Ambos hemisferios cerebrales están comprometidos desde el inicio de la crisis. La descarga cerebral anormal es bilateral. La manifestación clínica es diversa y siempre la crisis se inicia con alteración o compromiso de conciencia en el paciente. Las manifestaciones más comunes son:
En estas crisis no es posible definir claramente si se compromete solamente uno o ambos hemisferios cerebrales.
Se habla de síndromes epilépticos cuando existe un trastorno epiléptico con un conjunto de síntomas y signos característicos, tales como la edad de aparición de las crisis, tipo de crisis, hallazgos en el EEG, en la neuroradiología, respuesta al tratamiento, pronóstico a largo plazo. Poder clasificar una epilepsia como un síndrome epiléptico determinado permite muchas veces establecer la medicación a usar y la respuesta esperada al tratamiento. Ejemplos de síndromes epilépticos más frecuentes:
Es un síndrome epiléptico benigno que se presenta entre los 3-13 años de edad con un máximo entre los 5-10 años de vida. Representan alrededor de un 15% de las epilepsias del niño. Las crisis comprometen un lado del rostro, lengua, hay dificultad para tragar la saliva y para hablar. Suelen presentarse 1-2 horas después de iniciado el sueño o momentos antes de despertar y la mayoría de los casos tiene sólo unas pocas crisis. Sin embargo, a veces las crisis se generalizan y pueden confundirse con crisis tónico-clónicas del sueño. En cerca de un 20% de los casos las crisis pueden presentarse de día. El examen neurológico es normal. Puede haber mayor porcentaje de problemas de aprendizaje escolar. Invariablemente mejoran con la adolescencia. El EEG típico es con un ritmo de base normal y espigas en las regiones centrotemporales del cerebro. Alrededor de un 40% de los casos tienen historia familiar de epilepsia.
Se presenta entre los 3-11 años de edad con una mayor frecuencia relativa a los 5-6 años. Constituyen un 5-10% de las epilepsias en el niño. Las crisis se caracterizan por episodios de desconexión de 5-15 segundos con interrupción de la actividad en curso, mirada fija y a veces leve parpadeo o algunos automatismos de chupeteo o algunos movimientos como abrochar. Son de comienzo y final súbito, existe amnesia del evento (no recuerdan), aunque con el tiempo los niños se dan cuenta que se desconectan. Se repiten muchas veces al día y son de predominio matinal. Hay antecedentes familiares de epilepsia hasta en un 40% de los casos. El examen neurológico es normal y el EEG muestra un ritmo de base normal y durante las crisis aparecen descargas generalizadas de espiga onda lenta a 3 hertz por segundos. Es de excelente pronóstico, sin embargo, pueden a veces asociarse a crisis convulsivas tónico-clónica generalizadas.
Constituye alrededor de un 7% de los casos de epilepsia. Se presenta alrededor del inicio de la pubertad. Las crisis son matinales y coexisten sacudidas mioclónicas del despertar, que típicamente provocan caída de objetos de las manos (taza, cepillo dental). Es como un golpe de corriente único. Se asocian crisis tónico-clónicas generalizadas particularmente cuando hay privación de sueño. Pueden existir también ausencias y crisis fotosensibles como las provocadas por la televisión, videos juegos o luces estroboscópicas (Discoteca). Existe un elevado porcentaje de casos con antecedentes familiares de epilepsia. El examen neurológico es normal. El EEG se caracteriza por registro basal normal con descargas generalizadas de puntas y polipuntas ondas lentas visibles con mayor facilidad durante el sueño. Responde muy bien a los FAE, pero tiene invariablemente tendencia a la recaída al suspenderlos. Además, implica restricciones en el estilo de vida del adolescente y adulto joven en cuanto al riesgo de recaída que implican las trasnochadas y el alcohol.
Infrecuente pero severo. Se presenta en lactantes entre 3 y 18 meses de edad con un máximo alrededor de los 6 meses de vida. Las crisis consisten en flexión de la cabeza y un movimiento de abrazo que a veces asocia llanto, dura 1-2 segundos y se repite varias veces en salvas (espasmos). Se observa especialmente al quedarse dormido o al despertar y no asocian decaimiento post crisis. El síndrome de West tiene una triada clásica constituida por los espasmos, atraso o detención del desarrollo psicomotor y un patrón eléctrico muy anormal en el EEG que se denomina Hipsarritmia. No menos de un 80% de los casos son sintomáticos y su pronóstico es malo en cuanto a la normalidad del desarrollo psicomotor y por la posibilidad de continuarse después de los 2 años de edad, como una epilepsia sintomática multifocal o transformarse en un síndrome de Lennox Gastaut. Su tratamiento más eficaz está constituido por la farmacoterapia hormonal con la hormona adrenocórticotropa (ACTH), asociada a un antiepiléptico.
Corresponde a una forma poco frecuente de crisis mioclónicas, de inicio entre los 4 meses y 3 años. Se caracteriza por pequeños saltos musculares o mioclonías y el EEG presenta una actividad eléctrica que coincide con los saltos musculares.
No se conoce hasta ahora su causa y el interés de su diferencia con otras formas de crisis mioclónicas, radica en la benignidad de su pronóstico. Es mucho más frecuente en varones y tiene una excelente respuesta a los fármacos anticonvulsivantes
Constituye la epilepsia focal más frecuente. Las crisis se originan en las estructuras profundas del lóbulo temporal. La expresión clínica de las crisis es muy variada y son principalmente crisis focales con compromiso de consciencia e incluye sensaciones como una molestia epigástrica, sensaciones de temor, despersonalización u otras difíciles de describir por los pacientes. Luego pueden aparecer automatismos que consisten en movimientos sin un propósito: de boca, lengua, mano, marcha y la posibilidad de generalización secundaria con la aparición de una crisis tónico-clónica. Este tipo de crisis suele ser de difícil control con los fármacos antiepilépticos (FAE), especialmente si se trata de una esclerosis mesial temporal (atrofia de la zona interna de este lóbulo), situación en el cual la remisión completa (libre de crisis), no se observa en más de un 30% de los casos. Este tipo de epilepsia es el que tiene mejor respuesta al tratamiento quirúrgico. El EEG muestra lentitud focal temporal y presencia de espigas. Estos pacientes suelen asociar problemas importantes con la memoria.
Se trata de un lóbulo cerebral muy grande y con muchas conexiones y las manifestaciones clínicas son muy variadas y a veces rápidamente pueden presentar generalización secundaria. Las crisis frontales pueden ser nocturnas con conductas bizarras, crisis focales con compromiso de consciencia, crisis con giro de la cabeza u ojos, automatismos bilaterales complejos, detención del habla, etc. Las crisis frontales frecuentemente pueden ser confundidas con parasomnias (problemas de sueño) o pseudocrisis psicogénicas. El EEG dado el gran volumen de corteza cerebral frontal puede ser normal, inespecífico o incluso mostrar sólo hallazgos generalizados, lo cual hace más difícil el diagnóstico localizatorio de las crisis originadas en el lóbulo frontal.
Fue descrito por S. Ohtahara en 1967. Se trata de una enfermedad poco frecuente, que aparece en el periodo de recién nacido. Los niños con esta condición presentan espasmos tónicos, un tipo de crisis en las cuales se estiran y se ponen “duros”, detienen la respiración y se ponen morados. Estas crisis se presentan en grupos de 10 a 20, siendo seguidas a veces por llanto incontrolable.
Generalmente, son niños que nacieron gravemente asfixiados o con malformaciones del cerebro.
Al realizar un EEG, éste muestra un trazado más o menos característico con momentos en que casi no se ve la actividad eléctrica cerebral y algunos otros con descargas eléctricas anormales.
La tomografía cerebral (TAC) muestra grandes alteraciones. Es un cuadro muy grave. La mayoría de los niños mueren pronto y los que no lo hacen, quedan con secuelas neurológicas y con crisis convulsivas reiterativas. Muchos de estos casos evolucionan a un Síndrome de West.
Este síndrome debe su nombre a dos médicos especialistas en epilepsia, quienes fueron los primeros en describirlo. Se manifiesta con crisis de varios tipos y el EEG muestra alteraciones eléctricas específicas. Generalmente, se presenta después de los 2 años de edad, mayormente entre los 3 y 5 años. Aproximadamente la tercera parte de ellos derivan de un síndrome de West.
Los niños con este síndrome tienen discapacidad intelectual y presentan crisis que les hacen caer bruscamente al suelo, golpeándose e hiriéndose la cabeza y el rostro. Por eso se recomienda que estos pacientes usen siempre un casco como protección.
Estas crisis se denominan atónicas, o drop-attack (en inglés), y se acompañan de otras, como las tónicas, en las que al dormir los pacientes presentan posturas de extensión total, se ponen “duros y extendidos”, o si están despiertos extienden bruscamente la cabeza hacia atrás, con un parpadeo de ojos muy rápido.
También tienen crisis llamadas de ausencia compleja, en las que el niño se desconecta momentáneamente del ambiente presentando, además, movimientos automáticos de boca (como masticar o succionar) o de manos (como si se estuvieran abotonando la ropa).
Las causas son muy parecidas a las del síndrome de West: previas al nacimiento u originadas durante o después del mismo. En algunos casos, la aparición de este síndrome se debe a un status epiléptico o surge como secuela de una hemorragia cerebral. (Un status epiléptico es una situación en la que el niño tiene una crisis de más de treinta minutos de duración o muchas crisis seguidas en un lapso corto de tiempo, intervalo durante el cual permanece sin recuperar la conciencia).
Generalmente, se usan muchos fármacos para intentar controlar las crisis. Sin embargo, esto es muy complicado, pues son de difícil manejo, por lo que el niño termina tomando 3-4 o 5 fármacos con efectos no deseados, como sueño durante el día o a veces más crisis, sin encontrar alivio para su problema total. Últimamente, se ha vuelto a utilizar la dieta cetogénica como tratamiento para controlar las crisis, obteniéndose resultados positivos. En algunos casos es posible realizar cirugía.
La mayoría de estos niños no consigue mejorar su déficit cognitivo, aún con un buen control de crisis. En los pacientes con mal control, se produce una disminución progresiva de las condiciones intelectuales.
Esta es una forma poco común en la que evolucionan las llamadas convulsiones febriles o crisis febriles. A su vez, las convulsiones febriles pueden ser típicas o atípicas; siendo las atípicas aquellas infrecuentes desde su inicio, ya sea porque tienen una forma diferente, una mayor duración o porque se repiten de manera constante.
La historia típica es la de un niño que recibe su primera, a veces segunda, dosis de vacuna DPT y horas después, además de fiebre, presenta una crisis que es generalmente parcial (de un solo lado del cuerpo o de una sola extremidad), que dura más de 15 minutos o que se repite varias veces el mismo día o en días sucesivos.
De ahí en adelante, cada vez que tiene fiebre, las crisis tienen más o menos las mismas características hasta que, un día, empieza a presentar crisis sin fiebre. Bordeando los dos años, aparecen crisis mioclónicas (como sustos) y luego crisis focales con compromiso de conciencia, en las que el paciente se queda “pegado al vacío” con mirada fija y acciones automáticas. Aunque es un síndrome de inicio de niños pequeños y lactantes, puede prolongarse por muchos años.
Al inicio del cuadro, los niños son intelectualmente normales, pero, con el paso de los años, van perdiendo su desarrollo psicomotor y disminuyendo su capacidad intelectual. Adicionalmente, las crisis se vuelven cada vez más difíciles de tratar y muchos pueden desarrollar problemas de equilibrio. Aún en el grupo de niños que consigue quedar sin crisis, (alrededor del 40%) el compromiso intelectual existe y es progresivo.
Este síndrome no tiene una causa conocida, tiene que ver con la condición genética del paciente y se sospecha frente a la presentación de crisis febriles repetidas e inusuales. El EEG no es característico, pero ayuda en el diagnóstico al encontrar determinadas trazados eléctricos anormales.
Este síndrome debe su nombre a un cirujano canadiense que lo describió por primera vez y en su evolución es realmente catastrófico e, incluso, mortal. Se trata de una epilepsia focal (afecta a un miembro superior o parte de él) y continua, es decir, presenta crisis que no cesan ni de día ni de noche, pudiendo durar meses.
Afortunadamente, es de rara ocurrencia y suele presentarse antes de los diez años de vida, en un niño hasta entonces normal. Se producen crisis de tipo focal motor o con movimientos de grupos musculares de la mano, el antebrazo o todo el brazo y que no dejan descansar al paciente, quien está totalmente despierto.
Cuando se describió por primera vez, se asoció a una forma rara de encefalitis localizada (inflamación viral de una zona del cerebro), pero luego se ha descrito asociada a enfermedades metabólicas raras. En algunos pacientes no fue posible encontrar una causa clara.

La epilepsia es una afección crónica del Sistema Nervioso Central que se manifiesta en forma de crisis inesperadas y espontáneas, debido a actividad eléctrica excesiva de un grupo de neuronas del cerebro.
Todos podemos sufrir una crisis, aún sin tener antecedentes previos de epilepsia. Tener información sobre los pasos a seguir es clave a la hora de asistir a quien la experimenta.
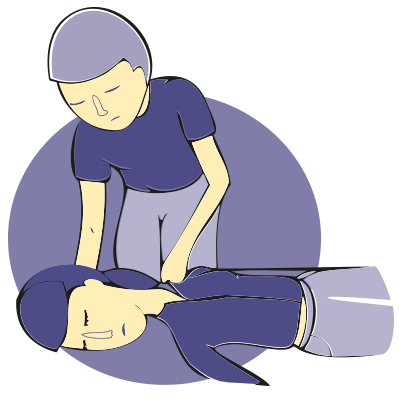
1.- Mantén la calma.
2.- No coloques nada dentro de la boca de la persona afectada, ni trates de abrirla a la fuerza. La lengua no se traga.
3.- No intentes sujetar ni reanimar a la persona. La mayoría de las crisis son breves y se produce recuperación espontánea.
4.- Protege la cabeza de posibles golpes poniendo ropa o un cojín debajo de ella.
5.- Recuesta a la persona suavemente en el suelo y despeja el área cercana para que no se golpee.
6.- Ponla de lado, especialmente su cabeza, para que pueda respirar mejor.
7.- Toma la hora por reloj: si la crisis dura menos de 5 minutos no es necesario trasladar a la persona a un servicio de urgencia.
Luego de la crisis, puede que la persona entre en un estado de sueño profundo, conocido como postictal. Si es así, déjala descansar hasta que se recupere completamente.


El diagnóstico de las epilepsias es fundamentalmente clínico, se realiza en base a un acabado relato de los síntomas que realiza el paciente (y/o los testigos de las crisis) y teniendo en cuenta los factores de riesgo de Epilepsia; además de los posibles factores precipitantes de crisis epilépticas. Se requiere, además, evaluar el desarrollo psicomotor e intelectual del niño, adolescente o adulto y realizar un examen físico y neurológico completo.
La Liga internacional contra la Epilepsia (ILAE), el año 2014, establece los siguientes criterios para hacer el diagnóstico de epilepsia:
Para complementar el diagnóstico, el médico solicitará un Electroencefalograma e imágenes del cerebro (neuroimágenes: scanner, TAC, resonancia, etc.), entre otros.
El electroencefalograma (EEG) es el registro gráfico de la actividad eléctrica producida por las neuronas en nuestro cerebro. El EEG durante el diagnóstico permite:
Para realizar el EEG se ponen unos electrodos en el cuero cabelludo y se conectan a una máquina, que registra la actividad eléctrica del momento. Este examen dura 20 minutos, pero, según indicación médica, puede hacerse un registro más prolongado (desde 1 hora, hasta 5 días de manera ambulatoria).
Durante la realización del estudio la persona debe permanecer acostada, con los ojos cerrados, si bien se le ordenará la apertura y el cierre intermitente de los párpados para observar los cambios que se producen en el registro. También se practicará la llamada hiperventilación, consistente en una serie de respiraciones profundas y continuadas, lo cual modifica la acidez de la sangre (ph sanguíneo) hacia el lado alcalino. Durante el registro se efectúa también la estimulación luminosa o fótica a frecuencias variables (fotoestimulación).
El EEG no hace el diagnóstico de Epilepsia en ausencia de síntomas (crisis), ya que las descargas epileptiformes que se pueden registrar no son exclusivas de las Epilepsias. Entonces, el EEG puede ser altamente específico para el diagnóstico de Epilepsia, pero solamente cuando existan crisis. Todo paciente con epilepsia debe hacerse un EEG en su evaluación inicial.
El EEG solo registra la actividad eléctrica de la zona del cerebro en contacto con los electrodos, si la persona tiene una epilepsia con crisis de sintomatología muy específica de un área del cerebro que no está siendo registrado por los electrodos, es probable que el examen salga informado como NORMAL lo cual no elimina la posibilidad de que ese paciente tenga epilepsia; es el médico quien debe hacer el juicio-diagnóstico correspondiente.
Por otra parte, cuando se pide un EEG por otra patología (cefalea, déficit atencional, autismo u otro) y éste sale con resultado alterado; esto NO significa que la persona tiene epilepsia en ausencia de crisis epilépticas. Para tener epilepsia siempre debe haber crisis epilépticas.
Una vez hecho el diagnóstico he indicado el tratamiento el EEG nos sirve para:
Son fundamentales para clasificar las epilepsias según etiologías (causas). La Resonancia Nuclear Magnética de Cerebro (RNM) es el “standard de oro”, pero debe realizarse con un protocolo especial para epilepsia. Permite diagnosticar epilepsias secundarias a malformaciones cerebrales, alteración de la migración neuronal, tumores y/o secuelas de enfermedades infecciosas (meningitis, encefalitis).
Ambos hemisferios cerebrales están comprometidos desde el inicio de la crisis. La descarga cerebral anormal es bilateral. La manifestación clínica es diversa y siempre la crisis se inicia con alteración o compromiso de conciencia en el paciente. Las manifestaciones más comunes son:
El objetivo del tratamiento para las epilepsias es mejorar la calidad de vida del paciente y su familia, esto se basa en tres pilares fundamentales:
1.- Fármacos antiepilépticos: adecuado control de crisis
2.- Medidas de autocuidado-automanejo: promover el automanejo de la enfermedad y evitar factores gatillantes de crisis.
3.- Manejo del Entorno social: Evitar el estigma y promover la inclusión
Los fármacos antiepilépticos (FAEs) son medicamentos que actúan a nivel cerebral, evitando que la crisis epiléptica se genere o se propague desde su foco de origen. El objetivo básico e inicial del tratamiento farmacológico, independientemente de la edad del paciente, será intentar la libertad o control de sus crisis epilépticas. Esto es fundamental para el óptimo funcionamiento cognitivo, mental y social del paciente.
Existen fármacos clásicos, algunos muy antiguos y otros nuevos. Su eficacia es similar, y la principal diferencia entre los medicamentos es que los fármacos nuevos tienen menos efectos adversos o toxicidad; pero su costo económico es significativamente más alto.
Es importante destacar que cada tratamiento es a la medida del paciente, tomando en cuenta su edad, sexo, escolaridad o actividad laboral y una adecuada clasificación del tipo de crisis y del Síndrome Epiléptico (cuando sea posible precisarlo). Además, siempre se debe considerar atenuar otras condiciones asociadas a la enfermedad o comorbilidades que pueda presentar el paciente. El tratamiento debe iniciarse cuando existe certeza y seguridad de que el diagnóstico corresponde a Epilepsia.
Siempre se inicia el tratamiento con MONOTERAPIA (un FAE) y la indicación debe tomar en cuenta: la clasificación del Síndrome Epiléptico, patologías asociadas, tolerabilidad del fármaco y seguridad del fármaco a largo plazo.
Si el primer FAE es mal tolerado, se usará un segundo FAE y si éste también fracasa en el control de las crisis se realizará una combinación de 2 FAEs, o, a veces de 3 FAES, en este caso hablaremos de un tratamiento con POLITERAPIA.
Si las crisis no se logran controlar con fármacos, el médico planteará el diagnóstico de Epilepsia Refractaria resistente a fármacos. En este caso se considerará evaluar y plantear otros tipos de tratamientos además del farmacológico, como:
De acuerdo a las particularidades que conlleva la epilepsia; características de la enfermedad, tipo de crisis, diagnóstico, tratamiento, entre otras; podemos plantear a una persona con diagnóstico de epilepsia, un plan general de automanejo de su enfermedad, este plan puede variar paciente a paciente dependiendo del tipo de epilepsia, crisis y tratamiento para cada uno:
1.- Registro de crisis:
Llevar un calendario detallado de las crisis registrando los días y horarios en que éstas se produzcan, ayuda al paciente a:
2.- Ingesta de medicamentos, en horarios y dosis indicados por el médico.
Los horarios no deben entorpecer el desarrollo de las actividades cotidianas, por ejemplo: debe tomar el medicamento antes de dormir y no tiene que despertarse a las 23 horas (si se queda dormido a las 21 horas) para seguir un horario estricto.
Buscando también el desarrollo de la autonomía, es recomendable el uso de un pastillero; dispositivo en el cual se cargan las dosis de medicamentos para la semana (mañana y tarde) y se toman de acuerdo al horario y días indicados. Con este método es fácil recordar si se tomó o no la dosis, basta mirar el cajón del pastillero con el día y horario correspondiente, si los medicamentos están ahí, no se los ha tomado y si no están ya lo tomó; evitando así duplicidad en la administración y olvidos de la terapia, ya que es más fácil tener el pastillero a mano, en el velador, cartera, mochila, etc.
El tratamiento con fármacos es por un periodo prolongado de tiempo (meses y/o años), por lo tanto, siempre debe mantener un control regular con su médico tratante, para el manejo de la Epilepsia y control de los posibles efectos adversos de los fármacos sobre el organismo.
3.- Higiene de sueño:
En las personas con epilepsia, la falta de sueño en horas o no tener un sueño reparador, es un gatillante de crisis; por lo tanto, cuidar las horas de sueño y dormir lo necesario, según la edad, es una medida de Automanejo fundamental para el adecuado control de las crisis.
4.- Manejo de las crisis epilépticas:
Esta es una medida de automanejo que debe conocer el paciente y todo su entorno, familiar y social. Es importante reconocer el tipo de crisis que tiene la persona (focal o generalizada) y estar preparados para prestar ayuda en caso que presente una crisis. (QUE HACER EN CASO DE CRISIS).
5.- Actividad física:
La limitación en la realización de actividad física estará condicionada a cada persona en particular, dependiendo específicamente de la actividad física que realiza o desee realizar, el tipo de Epilepsia que tiene y el control de sus crisis. La recomendación general es la siguiente
6.- Vida sexual y fertilidad:
Este tema se trata en profundidad en el capítulo de Epilepsia y mujer.
7.- Manejo factores gatillantes de crisis:
En la persona con epilepsia es importante reconocer su tipo de crisis y diferenciarla del resto de los eventos que pueden ocurrir a cualquier persona; además qué cosas del ambiente pueden ser precursores de crisis y cómo manejarlos para llevar una vida normal con el menor número de restricciones, respecto a una persona sin epilepsia. Dentro de los factores gatillantes de crisis se reconocen los siguientes:
Además, Como medidas de autocuidado- automanejo generales (no solo para las personas con epilepsia) no está de más recordar:
Se recomienda una alimentación saludable de acuerdo a su peso, talla y actividad física. Esto influye directamente en las dosis y respuesta de los fármacos para el control de la patología.
La Epilepsia es una enfermedad neurológica crónica milenaria, y la más frecuente en el mundo. A través de los siglos y de la mano de la ciencia, se han producido grandes avances en lo relativo a diagnóstico y tratamiento para esta patología. No obstante, un campo poco explorado aún, que requiere de gran atención y desarrollo e intervenciones profundas es el que se refiere a los aspectos psicosociales implicados en la Epilepsia, es decir, aquellos que se refieren a la interacción social del paciente, o cómo éste se vincula con la sociedad.
Las personas con epilepsia a lo largo de la historia han tenido dificultades en su participación social, a raíz del estigma que ha acompañado esta enfermedad desde sus orígenes. Muchas veces las consecuencias más negativas para la calidad de vida del paciente y su familia provienen de la mala reacción del entorno, por el rechazo y la discriminación social que con frecuencia viven algunas personas, siendo esto más perturbador incluso que las propias crisis epilépticas.
A nivel psicosocial, y según grupos etarios, el impacto psicosocial de la epilepsia se manifiesta de la siguiente manera:
Al parecer esta enfermedad también afecta la vida social de la persona con epilepsia, siendo más difícil su desempeño social, las relaciones afectivas, la vida en pareja e incluso la consolidación de un proyecto familiar que involucre matrimonio e hijos.
Se debe intentar que el paciente y su familia lleven una vida normal, con el menor número posible de restricciones. Sabemos que producto del diagnóstico y del tratamiento muchas veces será necesario implementar algunos cambios en el estilo de vida, y la familia resentirá además el peso del estigma de esta enfermedad con consecuencias negativas a nivel psicosocial.
Es importante fomentar el autocuidado del paciente y de su cuidador; entregar información acerca de las precauciones que la persona debe adquirir y la forma en la que debe seguir adecuadamente su tratamiento, con el apoyo y soporte de su familia.
Por otro lado, el manejo del ambiente y la educación de la comunidad para disminuir el estigma y favorecer la inclusión de las personas con epilepsia es vital y debe ser parte de las intervenciones que se realicen en pos de asegurar la calidad de vida de las personas con epilepsia. De esta forma, se hace necesario implementar y mantener el desarrollo de intervenciones psicosociales y educacionales, aspectos imprescindibles para el éxito del tratamiento.

Son crisis epilépticas de tipo convulsivas que ocurren en el lactante (de 1 a 24 meses) o preescolar (de 2 a 5 años), usualmente entre los 6 meses y 5 años, asociadas a fiebre, sin signos de infección intracraneana. Tampoco debe haber una enfermedad neurológica aguda. Se observan en un 3 a 5% de la población general, constituyéndose, por lo tanto, en el episodio convulsivo más común en los menores de 5 años. En un tercio de los casos son recurrentes, o sea, se presentan más de una vez. Tienen un carácter hereditario. No son Epilepsias y no requieren tratamiento, pero tienen un riesgo de epilepsia futura de 2 – 4 %, que es un riesgo algo mayor que la población general.
Las manifestaciones clínicas de las epilepsias de la niñez son muy variadas en relación a las del adulto, ya que el grado particular de maduración cerebral determina la capacidad para elaborar semiología (síntomas y signos) y la posibilidad de que el niño pueda relatar sus síntomas.
En niños menores de 5 años predominan algunas crisis generalizadas: tónicas, clónicas, hipomotoras y espasmos epilépticos. Muchas crisis de inicio focal se generalizan muy rápidamente pareciendo generalizadas, sin detectarse su inicio focal.
Existen muchos tipos de crisis que sólo se presentan a determinadas edades. Ej.: espasmos infantiles, crisis febriles, crisis de ausencia.
Existen 2 grandes grupos de etiologías (causas) que generan epilepsia en este rango de edad:
Son factores de riesgo en el niño como causales de epilepsia: Parálisis cerebral. déficit intelectual, infecciones del Sistema Nervioso Cental (SNC), trauma SNC, crisis febriles y algunos factores genéticos.
Se caracterizan por presentar crisis muy frecuentes y deterioro cognitivo y de conducta en el niño. La presencia de discapacidad intelectual refleja que las EE se originan durante el desarrollo neuronal, distorsionándolo, de modo que, incluso si las crisis pueden controlarse utilizando los fármacos antiepilépticos disponibles, el deterioro cognitivo no se puede detener con los actuales tratamientos. Esta es la razón por la cual las EE se consideran enfermedades neurodegenerativas y del neuro-desarrollo al mismo tiempo.
En el caso de niños y adolescentes con Epilepsia el tratamiento no es distinto al tratamiento general de esta enfermedad, consistiendo fundamentalmente en prescripción de FAES (fármacos antiepilépticos), instauración de conductas de autocuidado y control de los factores de riegos que son precipitantes de crisis.
La clasificación del tipo de epilepsia es clave porque algunos síndromes epilépticos y encefalopatías epilépticas responden a terapias específicas, que no se usan en la vida adulta (Ej ACTH, corticoides y vigabatrina, por ejemplo, en el Sd de West)
La epilepsia puede impactar negativamente la autoestima y el desarrollo de la identidad del adolescente; al tener que aceptar y asumir una enfermedad crónica como la epilepsia, que además es estigmatizante. Esto puede provocar sentimientos de frustración, vergüenza, angustia e inseguridad y también algunas dificultades conductuales para aceptar la enfermedad, o lograr la adaptación del joven y su entorno a un estilo de vida acorde a lo que requiere su tratamiento. Será necesario a veces desarrollar espacios de trabajo terapéutico y apoyarlo en la elaboración de estrategias de afrontamiento. En esta tarea son factores protectores el desempeño escolar o educacional y características de la familia.
Para el equipo de salud la gran tarea es entender las necesidades del adolescente, contenerlo, educarlo y apoyarlo para lograr su adherencia al tratamiento.
La duración del tratamiento con FAEs depende del tipo de epilepsia. Se considera un tiempo razonable de tratamiento, para muchas epilepsias de estos síndromes (no lesionales), de 2-3 años sin crisis. Pero hay otras epilepsias como la Mioclónica juvenil, que responde muy bien a la terapia farmacológica, pero que estará activa gran parte de la vida del paciente, lo que significa que necesitará tomar medicamentos por más tiempo.
En epilepsias de debut muy temprano, con grandes malformaciones de un hemisferio cerebral se debe plantear muy precozmente una cirugía de epilepsia como tratamiento paliativo (detener la frecuencia y severidad de las crisis).
El pronóstico de la Epilepsia en niños y adolescentes, depende fundamentalmente de la forma clínica:
Las comorbilidades más frecuentes son el Trastorno por Déficit de Atención y trastornos de la esfera emocional: trastornos por ansiedad y Trastornos de ánimo .
Epilepsia y Déficit de Atención (TDA): El TDA es 2.5 veces más frecuentemente en niños y adolescentes con epilepsia que en población general. La asociación ha sido atribuida a la propia epilepsia, a la anomalía cerebral de base o al tratamiento, aunque ha sido difícil determinar qué ocurre primero. Se debe pesquisar en forma dirigida ante una disminución de rendimientos en tareas de aprendizaje. El tratamiento con psicoestimulantes, asociados a los fármacos antiepilépticos (FAE), es una medida de tratamiento básica y segura en estos pacientes.
Trastornos de Ansiedad: El trastorno de ansiedad podemos caracterizarlo como una sensación negativa a algo que sucederá en el futuro (por ejemplo: frente a un examen o una prueba en el colegio o al tener que tomar alguna decisión afectiva). Es una señal de alerta frente a un posible daño y que suele generar angustia y tensión. Lo impredecible de las crisis genera inseguridad y vergüenza en el adolescente y es una fuente permanente de ansiedad. Las manifestaciones clínicas son variadas e incluyen dificultades de concentración, inhibición social, trastornos del sueño y síntomas de somatización (cefalea, dolor abdominal, dolores musculares, etc). Una alta frecuencia de crisis se asocia significativamente a baja autoestima y mayores niveles de ansiedad.
Trastornos de ánimo: Los trastornos del ánimo son frecuentes en adultos con epilepsia y han sido poco descritos en niños, pero constituyen una comorbilidad psiquiátrica frecuente. Se confunde frecuentemente con síntomas de depresión, trastornos conductuales y efectos secundarios de FAEs en niños y adolescentes con epilepsia. La depresión y la epilepsia pueden compartir mecanismos causales comunes que facilitan la ocurrencia de una en la presencia de la otra.
La depresión se presenta como un conjunto de síntomas de predominio afectivo (tristeza patológica, apatía, anhedonia, desesperanza, decaimiento, irritabilidad, sensación subjetiva de malestar e impotencia frente a las exigencias de la vida) entre otros síntomas.
Son predictores de depresión en niños y adolescentes:
A esta edad, la falta de información y desconocimiento sobre la epilepsia se relaciona con mayor riesgo de depresión, baja autoestima y ansiedad social.
Tanto los trastornos ansiosos como los que afectan al ánimo suelen interferir en las relaciones del niño y adolescente consigo mismo y con el entorno, por ello se recomienda tratamiento psicológico acompañado de evaluación psiquiátrica y frecuentemente terapia farmacológica específica. Cabe destacar, que la gran mayoría de las dificultades de adaptación social y de identidad se vinculan más bien a las relaciones con los padres y amigos que a la gravedad misma de la epilepsia.
La adolescencia tiene sus propias problemáticas y estos trastornos emocionales constituyen una importante comorbilidad en epilepsia por lo que requiere de estar atentos para lograr detectarlos oportunamente y realizar los tratamientos pertinentes.

Hasta un 50% de las mujeres con epilepsia tendrían trastornos del ciclo menstrual y mayor frecuencia de ciclos anovulatorios. Estos fenómenos se presentan tanto en mujeres con epilepsias focales como generalizadas.
La anovulación es más frecuente en pacientes con epilepsia de lóbulo temporal y poli terapia (más de un fármaco antiepiléptico)
Principales signos clínicos de Trastornos Endocrinos asociados a Infertilidad:
Se sospecha, cuando en la mujer existe Hiperandrogenismo con un estado de anovulación crónica en mujeres sin otras patologías específicas de las glándulas adrenales o pituitarias. Es decir, la definición de Síndrome de ovario poli quístico (SOP) no requiere de la presencia de quistes en los ovarios (es un fenómeno hormonal). Un 20% de las mujeres sí presenta ovarios poliquísticos.
El SOP es frecuente en la mujer en edad fértil (prevalencia de 2 a 5%) y es la principal causa de infertilidad anovulatoria.
El diagnóstico se realiza si se encuentran los siguientes síntomas:
Los fármacos antiepilépticos pueden tener efectos sobre la función endocrina y reproductiva, los que se manifiestan:
Las hormonas sexuales femeninas pueden influenciar la facilidad y la capacidad que tiene el cerebro para producir crisis epilépticas. Así, por ejemplo, un tipo de hormona conocida como estrógeno facilita la aparición de crisis epilépticas mientras que otro tipo de hormona, la progesterona, dificulta la aparición de crisis epilépticas. Existe un predominio de uno u otro tipo de hormona durante el ciclo menstrual, lo que explica que en algunas mujeres las crisis aparezcan o aumenten con relación a distintas etapas del ciclo. A este patrón de las crisis se lo conoce como epilepsia catamenial.
El diagnóstico de epilepsia catamenial requiere de un registro cuidadoso en un calendario de crisis (anotar día a día, el número y tipo de crisis) de varios ciclos menstruales, ya que, en la práctica clínica, es frecuente que se tenga la impresión de que las crisis se asocian a alguna etapa del ciclo menstrual, pero que esto no sea así. En el caso de que se confirme una epilepsia catamenial, existen varios medicamentos que pueden ser usados en forma intermitente para el control de las crisis, los que deben ser evaluados en conjunto con el médico.
En algunas mujeres la llegada de la menopausia podría producir un aumento de las crisis, especialmente en aquellas que se encuentren en tratamiento de sustitución con estrógenos.
Los anticonceptivos orales (ACOs) inducen la glucurunazión a nivel hepático, lo que puede inducir en el metabolismo de ciertos fármacos antiepilépticos (ejemplo: Lamotrigina, Oxcarbacepina (MHD), Acido Valproico).
Hay considerar que existe una importante variabilidad interindividual, cada mujer se comporta de manera diferente frente a la terapia, pero se recomienda evitar el uso de anticonceptivos orales (estrógenos) ya que tienen múltiples interacciones cruzadas con los FAEs o hacerlo bajo estricta supervisión médica.
Alternativas a los anticonceptivos orales:
Es necesario antes de comenzar cualquier terapia anticonceptiva, consultar a médico tratante (ginecólogo y neurólogo) quienes aconsejaran el mejor método según el fármaco antiepiléptico que esté usando y la realidad personal de cada mujer.
El mejor predictor de la frecuencia de crisis durante el embarazo parece ser la frecuencia de crisis en el año previo al embarazo. El aumento en la frecuencia de crisis ha sido atribuido a múltiples causas:
Durante el embarazo se presentan fluctuaciones en el nivel plasmático (disponibilidad en la sangre) de los fármacos antiepilépticos (FAEs) debido a alteraciones en:
Se estima que una reducción mayor de 35% de la disponibilidad total del fármaco en la sangre, podría resultar en un aumento de la frecuencia de crisis. Idealmente se debiera usar el nivel plasmático previo al embarazo para orientar los eventuales cambios en la dosificación.
Para establecer objetivamente el riesgo de malformaciones fetales (teratogénesis), asociado a la exposición a Fármacos antiepilépticos; basamos nuestras recomendaciones en tres grandes estudios mundiales prospectivos
A través de éstos estudios se puede concluir que, Las mujeres sin epilepsia tienen 2 a 4% de posibilidades de tener un hijo que presente algún tipo de malformación. En el caso de una mujer con epilepsia esta probabilidad se duplica alcanzando una cifra de 4 a 6%. Sin embargo, aún conserva una posibilidad de tener un hijo sano de 94 a 96% (comparado con un 96 a 98% de una mujer sin epilepsia). En la práctica, esta diferencia en el riesgo de malformaciones es mínima y resulta muy tranquilizadora para la mujer con epilepsia que está planificando un embarazo. El mayor riesgo de malformaciones en los hijos de madres con epilepsia se debe principalmente a los FAE y no a la enfermedad, pero POR NINGUN MOTIVO se debe suspender la terapia farmacológica, cuando la mujer sabe que está embarazada.
En general, no existe por el momento evidencia suficiente que permita identificar a uno o más FAE que se asocien a un grado mayor o menor de malformaciones del recién nacido. Sin embargo, sí es claro que el ácido valproico y la carbamazepina se asocian a un mayor riesgo de que el recién nacido presente una malformación grave del sistema nervioso conocida como espina bífida (1- 2% y 0,5 – 1 % respectivamente). Es por ello, que se tratará de evitar el uso de éstos fármacos si se está planificando un embarazo. Aún en el caso de estos medicamentos, rige la norma de no suspender el tratamiento ante el diagnóstico de un embarazo no planificado, ya que esta malformación se produce dentro de los primeros 28 días de gestación y por lo tanto ya está presente en el momento del diagnóstico de embarazo. Suspender el tratamiento en ese momento sólo le agrega un riesgo más al niño en gestación. Por otra parte, como la probabilidad de que la espina bífida se produzca es tan baja, la inmensa mayoría de estos niños es completamente normal.
El ácido fólico es una vitamina que disminuye en las mujeres que toman FAEs. Cuando una mujer se embaraza también disminuyen los niveles de ácido fólico en la sangre.
El ácido fólico ayuda a prevenir las malformaciones del recién nacido, especialmente aquellas más graves que comprometen al sistema nervioso.
El ácido fólico debe administrarse además de los FAE, en TODA mujer en edad fértil con epilepsia, incluyendo a las adolescentes y a las mujeres sin vida sexual activa.
Los FAE con baja unión a proteínas tales como Fenobarbital, Lamotrigina, Levetiracetam, Topiramato y Gabapentina pasan a la leche materna como para esperar efectos Clínicos. Varios estudios de series de casos no han reportado efectos adversos en la leche excepto por la sedación que pueden producir los barbitúricos y los benzodiacepinas en el lactante.
Esta no es una contraindicación para la Lactancia materna en mujeres que tomen FAES, pueden hacerlo con control regular de su neurólogo y pediatra del recién nacido. Por lo tanto, siempre se debe promover la lactancia cualquier sea el FAE con el que esté siendo tratada la paciente.
Se recomienda amamantar de costado o en presencia de un familiar.
La fenitoína, el fenobarbital y en menor medida la carbamazepina y el ácido valproico disminuyen el calcio de los huesos, aumentando el riesgo de osteoporosis (desmineralización de los huesos) y fracturas. Por lo tanto, las pacientes en tratamiento con estos FAE deben efectuar controles regulares de su calcemia, nivel plasmático de vitamina D y hormona paratiroídea, de acuerdo a las indicaciones del especialista. Se recomienda además la realización de un examen llamado densitometría ósea (cuyo propósito es medir el nivel de calcio en los huesos) al completar 5 años de tratamiento con algunos de los FAE previamente mencionados.
El efecto sobre la salud ósea de otros FAE de aparición más reciente no ha sido aún completamente evaluado, por lo que es necesario mantenerse atento en cuanto a la aparición de nueva información en esta área. En el caso de confirmarse una menor fijación de calcio o una osteoporosis, existen tratamientos disponibles sobre la base de administración de calcio, Vitamina D o fármacos para aumentar la fijación ósea de calcio.

Es la Comorbilidad psiquiátrica más frecuente en Epilepsia.
La Depresión se presenta con síntomas como: bajo ánimo, anhedonia, inhibición corporal y cognitiva, culpa y desvalorización de sí mismo, desesperanza, abulia, irritabilidad, alteración del sueño y apetito.
Tiene una prevalencia entre 30 y 63 % en pacientes con epilepsia, es más común en epilepsia del Lóbulo Temporal y existe alta tasa de suicidio en pacientes con epilepsia: 5 a 25 veces más alto en pacientes con epilepsia lóbulo temporal y depresión que población general.
Se asocia a Múltiples factores:
El tratamiento es principalmente con Antidepresivos, pero a pesar de la alta prevalencia de Depresión en Epilepsia, existe baja prescripción de antidepresivos en pacientes con Epilepsia, ya que aún permanece la creencia que éstos actúan como pro-convulsivantes.
Los antidepresivos son seguros para epilepsia en dosis terapéuticas, incluso cada vez existe más evidencia sobre posible efecto anticonvulsivante de los antidepresivos.
Mayores interacciones: paroxetina, fluoxetina, fluvoxamina.
Menores interacciones: Citalopram, escitalopram, Sertralina, Venlafaxina, Desvenlafaxina, Mirtazapina.
Otras consideraciones a evaluar:
Hay fármacos antiepilépticos que aumentan el riesgo de depresión: levetiracetam, Fenobarbital, Topiramato, Perampanel, lacosamida, benzodiacepinas. Y otros fármacos antiepilépticos que disminuyen el riesgo de depresión: lamotrigina, carbamazepina, ácido valproico, pregabalina, benzodiacepinas; por lo que el tratamiento DEBE ser indicado por el médico especialista (neurólogo y psiquiatra).

Concepto adulto mayor según OMS: Persona mayor de 60 años en países desarrollados, persona mayor 65 años en países subdesarrollados
Incidencia y prevalencia de epilepsia en adulto mayor es alta: el doble que población joven
La Epilepsia representa la 3° causa patología neurológica en este grupo etario
Entre los factores externos que pueden producir epilepsia en el adulto mayor, se encuentran:
El tratamiento, en estos casos, está orientado al control de las crisis a través del uso de FAEs y evitando la causa desencadenante.
Además, existen algunas enfermedades neurológicas que pueden producir epilepsia en los mayores de 60 años, como, por ejemplo:
Existe un retardo diagnostico que puede ir de días a años desde la primera consulta: Retardo diagnostico en promedio 1.7 años; Mayor retardo diagnóstico en mujeres con epilepsia focal; 50% no consulto de inmediato, demoró en promedio 9 meses en consultar.+
El objetivo básico del tratamiento de la epilepsia, independientemente de la edad del paciente, es el control de las crisis. En muchos casos, este objetivo se logra a través de la utilización de sólo un fármaco, mientras que, en otros, será necesario combinar más de uno para lograr un control adecuado.
Los FAEs utilizados en el tratamiento de adultos mayores, son esencialmente los mismos que se emplean con los otros grupos etarios. Sin embargo, un tratamiento adecuado deberá considerar las características propias del organismo de la persona (metabolismo y eliminación lenta de los tóxicos del cuerpo) y la frecuencia de la coexistencia de otras enfermedades, lo que, en muchos casos, obliga a poli-medicar al paciente.
Asimismo, es fundamental considerar los recursos económicos del paciente, de manera de evitar fracasos terapéuticos por abandono del tratamiento.

La epilepsia es una causa de consulta común en neurología veterinaria, ya que los animales también la pueden presentar. Corresponde a una condición cerebral crónica, que se caracteriza por la presentación de convulsiones (crisis o ataques epilépticos), que se repiten en el tiempo, con una frecuencia variable. La epilepsia corresponde a la manifestación clínica de diferentes patologías que afectan al cerebro, y no a una enfermedad en sí. Debido a esto, cuando se presentan crisis epilépticas es recomendado establecer un plan diagnóstico para encontrar la causa primaria, y no solo remitirse a dar drogas anticonvulsivantes.
– Epilepsia idiopática, que se basa en anormalidades genéticas que llevan a la presentación de crisis epilépticas, sin ninguna causa aparente.
– Epilepsia estructural, que ocurre secundario a enfermedades cerebrales como malformaciones congénitas, tumores, inflamación, trauma, entre otras.
Las crisis epilépticas, al igual que en los humanos, son la manifestación clínica transitoria de una alteración en la actividad cerebral, que pueden presentarse con actividad motora, conductual y/o autonómica. Probablemente la más conocida es la del tipo tónico-clónicas, en que el animal pierde el conocimiento, cae de lado, se muestra rígido y presenta movimientos de contracción en sus extremidades. Sin embargo, existen múltiples presentaciones dependiendo de la región del cerebro afectada, como por ejemplo sólo rigidez, cambio de conducta, movimientos masticatorios. Las crisis epilépticas se caracterizan por ser autolimitadas y durar menos de 2 minutos, aunque en ocasiones pueden existir una seguidilla de crisis (o un estado epiléptico), que es de larga duración y requiere atención de urgencia.
Para el diagnóstico, es fundamental que el animal sea evaluado por un médico veterinario y que se consideren diferentes aspectos, para poder determinar si es epilepsia idiopática o estructural, ya que ello depende el manejo terapéutico recomendado.
Habitualmente un perro o gato con epilepsia idiopática presenta todo normal, y sólo se ve afectado por las crisis epilépticas.
Uno de los manejos terapéuticos más común y efectivo, es la utilización de fármacos anticonvulsivantes. Estas son similares a las utilizadas en medicina humana, sin embargo, no todas son recomendadas para el uso en mascotas. El inicio y la elección de este tipo de medicamentos, se realiza considerando aspectos como la especie, frecuencia de las crisis, tipo de epilepsia, resultado de pruebas sanguíneas, entre otras.
En el caso de epilepsia estructural, además del uso de drogas anticonvulsivantes, se deben hacer manejos específicos de la causa primaria, que puede ir desde la prescripción de otros medicamentos hasta una cirugía.
El éxito de una terapia anticonvulsivante, está enfocado en la disminución en la frecuencia e intensidad de las crisis, ya que en la mayoría de los casos no desaparecen por completo.
Con un trabajo en conjunto entre el médico veterinario y los propietarios de mascotas con epilepsia, se puede entregar las condiciones más adecuadas para una buena calidad de vida de los pacientes y su familia.
Javier Green Lazo
Médico Veterinario
Director médico de SEDIVET.
Es una enfermedad neurológica crónica, en donde existe una alteración de la corteza cerebral que se caracteriza por la predisposición duradera a tener crisis epilépticas espontáneas. Y que trae consigo, además, una serie de repercusiones psicológicas y psicosociales.
Las crisis epilépticas se definen como la manifestación clínica de una actividad eléctrica anormal (descarga excesiva o cortocircuito) de nuestro cerebro. Se manifiestan por síntomas (lo que siente la persona) y signos (lo que podemos observar) paroxísticos (de inicio y término muy definido) y usualmente breves (transitorios).
Aquellas en las que la descarga cerebral anormal se inicia en forma simultánea (sincrónicamente) en todo el cerebro, y por consiguiente, existe pérdida de conciencia desde el inicio. Un ejemplo de ella es una crisis con un componente motor, la crisis tónico-clónica generalizada (la que conocemos como “convulsión”): la persona pierde el conocimiento, cae al suelo, hay rigidez muscular y luego sacudidas del cuerpo, bota una saliva espumosa, pudiendo morderse la lengua y orinarse.
Aquellas en que la descarga cerebral anormal se inicia en un área restringida del cerebro, pudiendo luego generalizarse. Inicialmente pueden darse sin alteración de conciencia y pueden o no acompañarse de movimientos anormales. Un ejemplo es una crisis focal sensitiva que se manifiesta por una sensación de hormigueo de brazo derecho que a veces se propaga a la mitad derecha de la cara sin alteración de conciencia.
En general, cuando hablamos de Epilepsia, se dice que la persona puede tener de 1 a 3 tipos de crisis y esto se debe a:
1.- El tipo de epilepsia que tenga y cómo ésta evoluciona en el tiempo
2.- La coexistencia de crisis focales y generalizadas.
No. El cerebro tiene mecanismos para protegerse de una crisis de epilepsia. Solo si la duración es más prolongada se puede producir daño neuronal, por ejemplo, cuando han transcurrido más de 30 minutos de iniciada la crisis. Por esto si la crisis supera los 5 minutos, se recomienda trasladar a Servicio de Urgencia o llamar una ambulancia.
Un conjunto de signos y síntomas definidos como epilépticos que configura entidades clínicas características con etiologías (causas) diferentes. Consideran elementos como, tipos de crisis, edad de comienzo, anomalías del EEG y neuroimagen, factores genéticos y otros. Determinan respuesta a algunos fármacos antiepilépticos y pronóstico. Ej. Síndrome de West.
No todas las Epilepsias pueden clasificarse en Síndromes.
La etiología o causas pueden ser variadas y diversas, y se pueden asociar a ciertas edades o grupos etarios algunas causas específicas.
A nivel mundial existen 5 causas frecuentes de epilepsia:
En la mayoría de los casos de Epilepsia, no hay una lesión en el cerebro demostrada que explique la razón de la enfermedad (hasta un 65% de las epilepsias diagnosticadas). A este tipo de epilepsias se les conoce como primarias, idiopáticas o determinadas genéticamente.
No. Una de cada de 10 personas puede presentar una crisis epiléptica a lo largo de su vida y no tener epilepsia, porque existen crisis epilépticas provocadas o denominadas crisis sintomáticas agudas.
Una crisis que se produce por una alteración aguda, transitoria y en general reversible del funcionamiento cerebral. Ej crisis febriles, hipoglicemia (falta de azúcar en la sangre), crisis provocada por ciertos fármacos, deprivación o intoxicación alcohólica, etc.
La definición actual exige al menos 1 de 3 requisitos para el diagnóstico:
El diagnóstico es clínico, idealmente debe hacerlo un neurólogo y se complementará con algunos exámenes, como el Electroencefalograma, pruebas de imágenes y estudios genéticos.
El electroencefalograma (EEG) es el registro gráfico de la actividad eléctrica producida por las neuronas de nuestro cerebro. Este examen puede ayudar a identificar una actividad eléctrica anormal y así corroborar y ayudar a precisar el tipo de epilepsia en una persona.
Todo paciente con epilepsia debe hacerse un EEG en su evaluación inicial.
El EEG solo registra la actividad eléctrica de la zona del cerebro en contacto con los electrodos. Si la persona tiene una epilepsia con crisis de sintomatología muy específica de un área del cerebro que no está siendo registrado por los electrodos, es probable que el examen salga informado como NORMAL lo cual no elimina la posibilidad de que ese paciente tenga epilepsia; es el médico quien debe hacer el juicio-diagnóstico correspondiente.
Por otra parte, cuando se pide un EEG por otra patología (cefalea, déficit atencional, autismo u otro) y éste sale alterado no se puede hacer el diagnóstico de Epilepsia. No existe epilepsia en una persona que no tenga crisis epilépticas.
La privación de sueño, es un gatillante de actividad y crisis epilépticas. Por lo tanto, si la persona no duerme, al momento de realizar el examen es posible registrar sueño y más probable detectar actividad epiléptica.
Para establecer la causa de la epilepsia en un paciente determinado, el médico puede solicitar distintos estudios que permitan tener imágenes de las diferentes estructuras del cerebro y detectar algunas anomalías que pueden ser el origen de la epilepsia. Para ello se puede solicitar una Tomografía axial computarizada (TAC o Scanner) y/o una Resonancia Magnética de cerebro, siendo esta última el examen de preferencia para descartar alguna malformación o daño a nivel cerebral.
Mejorar la calidad de vida del paciente y su familia.
Esto se basa en tres pilares fundamentales:
1.- Fármacos antiepilépticos: para lograr adecuado control de crisis
2.- Medidas de autocuidado: promover el automanejo de la enfermedad y evitar factores gatillantes de crisis.
3.- Manejo del Entorno social: evitar el estigma y promover la inclusión
Son medicamentos que disminuyen la frecuencia y severidad de las crisis en personas con epilepsia. Tratan los síntomas de las crisis, no la condición epiléptica de base. El resultado esperado es maximizar la calidad de vida del paciente con los menores efectos adversos posibles.
El propósito del tratamiento con medicamentos es el prevenir las crisis, no curarlas; por ello la medicación deber ser tomada por periodos largos de tiempo (meses o años). Lo que se quiere lograr con el tratamiento farmacológico es la libertad de crisis, no se curará la causa de la Epilepsia, pero se puede lograr que la persona esté sin crisis epilépticas pese a tener la enfermedad.
Un 70% de las personas diagnosticadas con Epilepsia tendrá un buen control de crisis con fármacos.
La duración del tratamiento depende del tipo de epilepsia y es el médico quien debe decidir el momento de retirar el tratamiento. Dependiendo del tipo de Epilepsia, frecuencia de crisis, entre otros factores.
En general se habla de 2 años de tratamiento para los niños y 5 años en los adultos; pero estos tiempos son arbitrarios, cada terapia se evalúa de manera particular médico-paciente, tipo de Epilepsia, calidad de vida y se decide si es pertinente o no dejar de usar los fármacos.
Si pudiéramos resumir diríamos que el autocuidado es el paraguas de actividades que realizamos los individuos para el cuidado de nuestra salud y el automanejo sería las actividades específicas que debemos modificar y/o agregar a nuestra rutina cuando aparece una enfermedad crónica.
De acuerdo a las características de las Epilepsias, tipo de crisis, diagnóstico, tratamiento, entre otras; podemos plantear a una persona un plan general de automanejo de su enfermedad. Este plan puede variar paciente a paciente dependiendo del tipo de epilepsia, crisis y tratamiento para cada uno.
Les llamamos gatillantes de crisis a aquellos factores o actividades de la vida diaria que pueden influir en que una crisis epiléptica aparezca, por ejemplo: falta de sueño, dormir poco, olvidar la dosis de medicamento, estrés, entre otros.
Esto puede ocurrir en un 30% de los pacientes diagnosticados con Epilepsia y se habla de Epilepsias refractarias o farmacorresistentes. El diagnóstico es médico, y si es el caso, se brindará al paciente y su familia todo el apoyo de un equipo multidisciplinario y según sea el caso se evaluarán otras alternativas de tratamiento, además de los fármacos (dieta cetogénica, cirugía, entre otros)
Actualmente sabemos que las personas con epilepsia no tienen restricciones en su alimentación: todas las comidas y bebidas están permitidas excepto el alcohol.
Sin embargo, hay ciertos alimentos o bebidas en los cuales recomendamos el consumo con cuidado por los efectos que tienen y cómo estos pueden afectar a las personas con epilepsia, por ejemplo: bebidas energéticas, potenciadores metabólicos (suplementos para deportistas), ginkgo biloba, anís estrellado, hierba de San juan, entre otros.
Siempre se debe mantener la calma, aunque sabemos que es un evento estresante y que nos provoca “miedo”.
1.- Evitar que la persona se golpee: tratar de recostarlo en el suelo y despejar el espacio alrededor.
2.- Poner algo blando bajo su cabeza (mochila, chaleco, cartera, etc.), lo que se tenga a mano.
3.- Tomar el tiempo, desde que se inicia el evento hasta que la persona recupera el tono muscular (deja de estar rígida). En el caso de crisis focales, hasta que responde.
4.- NO poner nada dentro de la boca (la lengua puede morderse, pero no es grave)
5.- Una vez que termina la crisis (el cuerpo deja de estar rígido), acostar de lado. Detener conteo de tiempo (habitualmente transcurrieron 1 a 2 minutos).
6.- Una vez de lado, la persona aún estará comprometida de conciencia (puede que no responda), pero NO ESTA CONVULSIONANDO. Hasta que recupere la conciencia pueden pasar 15 a 20 minutos. Nunca lo deje solo.
7.- Una vez despierte (recupere la conciencia), llame algún familiar o acompáñelo a su domicilio.